¿Qué determina la naturaleza y propiedades covalentes de una sustancia? 10 h


El enlace covalente es el resultado de la atracción entre los núcleos atómicos y pares de electrones compartidos entre ambos átomos. Eso se da fundamentalmente, cuando los átomos que se unen son no metales, y con alta electronegatividad por tanto.

El número de electrones que un átomo tiende a compartir está relacionado con la ya estudiada regla del octeto, por la cuál, los átomos tienden a rellenar su capa de valencia con 8 electrones, para asemejarse así a un gas noble. Así, un átomo como el flúor, con 7 electrones, sólo necesita 1 para completar el octeto, y por ello, comparte también 1; el carbono, con 4, necesita otros 4 y por ello comparte los 4 que tiene, y así progresivamente. Cada par de electrones compartido es un enlace, si se comparten dos pares, se denomina doble, y si tres pares, triple.

Las estructuras de Lewis son representaciones planas de los enlaces en la molécula, realizadas con la premisa de que debe completarse el octete electrónico para todos los átomos presentes.


La fortaleza de los enlaces va acorde con el número de pares de electrones compartidos, y así:
º > = > -
Como más pares significa una mayor atracción, la longitud del enlace sigue el orden inverso:
º < = < -


En ocasiones, los dos electrones compartidos los aporta un único átomo, como en el ion amonio, en ese caso, el enlace covalente se denomina dativo o coordinado, pero hay que resaltar que es un enlace covalente idéntico e indistinguible de los demás.


Las estructuras de Lewis son planas y no representan la geometría de las especies químicas, y ese es uno de los objetivos del estudio de las moléculas. Para predecir la geometría, usamos la Teoría de la Repulsión de los Pares de Electrones de la Capa de Valencia, (Valence Shell Electron Pair Repulsion, VSEPR). Según esta teoría, los pares de electrones que puedan existir alrededor de un átomo, se ubican los más alejados posible entre sí para minimizar las repulsiones entre ellos.
Los pares de electrones se dividen en dos grupos, aquellos que forman enlaces y que unen átomos, y los que no; los primeros se denominan pares de enlace (X) y los segundos, pares de no enlace (E). A efectos de la VSEPR, los enlaces múltiples cuentan como un solo par.
Una vez que hemos dividido los electrones en pares, la geometría básica está determinada, y luego la intensidad relativa de las repulsiones entre los distintos pares, la modificará ligeramente. Para ello se debe tener en cuenta que los pares de no enlace se repelen con más intensidad entre sí que con los de enlace, y éstos entre sí son los que menos se repelen: E-E>E-X>X-X (Ver tutorial en este ENLACE).


Prueba este SIMULADOR que aplica el modelo VSEPR a varias especies que luego a aparecen en la hoja de problemas.

La atracción por los pares de electrones puede ser desigual en función de la electronegatividad de cada átomo, y eso lleva a la aparición de polaridad, pues los electrones no se distribuyen homogéneamente entre ambos. El enlace será covalente puro si la diferencia de electronegatividad es 0, covalente polar si está entre 0 y 1.8, y si es mayor que 1.8, el enlace será iónico, no covalente.



Para que una molécula sea polar, se deben dar dos circunstancias: los enlaces deben ser polares, y además, los dipolos generados en cada uno no deben anularse entre sí geométricamente:



Todo lo que hemos visto hasta ahora en cuanto a enlace covalente, corresponde a sustancias que forman moléculas. Sin embargo, existen otra sustancias, cuyos átomos también se unen entre sí por enlaces covalentes, formando estructuras tridimensionales extendidas sin que existan moléculas. Estas sustancias, llamadas a veces compuestos covalentes gigantes o reticulares, tienen propiedades radicalmente diferentes a los compuestos covalentes moleculares, pues las de éstos viene determinada por la intensidad de las fuerzas intermoleculares que veremos más adelante. Algunas de estas sustancias covalentes reticulares son el diamante o el grafito (ambos son formas alotrópicas del carbono), el silicio, y el cuarzo.


En el diamante, el carbono está unido covalentemente a otros cuatro átomos de carbono mediante enlaces simples, resultando en una estructura tetraédrica con ángulos de 109.5º. En el grafito, los átomos se unen sólo a otros tres, en una estructura trigonal plana con ángulos de 120º, formando láminas que luego se unen entre sí mediante fuerzas de London. Existen además enlaces dobles deslocalizados en las láminas de un modo similar a lo que sucede en el benceno. El grafeno es básicamente igual que el grafito, ya que es una única de las láminas que forma del grafito. Finalmente, los fullerenos y nanotubos tienen una estructura en la que los átomos de carbono se unen a otros tres átomos en estructuras que pueden ser hexagonales o pentagonales, pero en cualquier caso, no planas; al igual que el grafito, ambos presentan electrones deslocalizados, y los ángulos de enlace son intermedios entre 109 y 120º. La diferencia entre ambos es que los nanotubos son redes covalentes, mientras que los fullerenos son moleculares, y por lo tanto, presentan entre ellos fuerzas de London, como las láminas de grafito.
Imagen Jmol del diamante
Imagen Jmol del fullereno
El silicio tiene la misma estructura tridimensional que el diamante, en tanto que la del cuarzo se puede visualizar como la unión por los vértices de tetraedros formados por una átomo de Si unido a cuatro átomos de O:


Los compuestos covalentes reticulares son muy duros (el diamante es la sustancia más dura de la Tierra), pero no son frágiles como los iónicos; tienen puntos de fusión muy altos, son aislantes térmicos y eléctricos (hay excepciones a esto, como el grafito, por ejemplo) y no suelen ser solubles en ningún tipo de disolvente.


Las propiedades de los compuestos covalentes moleculares está determinada por la fortaleza de las interacciones que se desarrollan entre sus moléculas. Las interacciones intermoleculares se pueden clasificar del siguiente modo:
- Puentes de hidrógeno
- Fuerzas de Van der Waals
- Dipolo-dipolo
- Dipolo-dipolo inducido
- Dipolo instantáneo-dipolo instantáneo o de London
Los puentes de hidrógeno aparecen cuando tenemos H unido a F, O ó N (los tres elementos más electronegativos). Lo que ocurre entonces es que, dado que el H es muy pequeño y tienen un solo electrón, y el otro átomo es muy electronegativo, y atrae fuertemente al par de enlace, se genera un dipolo muy intenso en la molécula que, consecuentemente genera una potente interacción entre las moléculas. Esta interacción es la responsable de las excepcionales propiedades del agua, el amoniaco y el fluoruro de hidrógeno; de la complementariedad entre las bases nitrogenadas de las hebras de ADN; de parte de la estructura secundaria de las proteínas; de parte de la reactividad de las enzimas, etc.


Quizás esta es la gráfica más usada para verificar el efecto de los puentes de hidrógeno en propiedades de las sustancias, concretamente el punto de ebullición. Es evidente que en fluoruro de hidrógeno, agua y amoniaco, el punto de ebullición es anómalo, debido a la presencia de los puentes de hidrógeno. Además, se puede entender el orden: el agua puede formar dos pares de puentes de hidrógeno, al tener dos hidrógenos y dos pares de no enlace, sin embargo, las otras dos sustancias solo pueden forma un par, al tener o solo un H o solo un par de no enlace. Y dentro de estos dos últimos, dado que el dipolo es más fuerte en fluoruro de hidrógeno que en amoniaco, por la mayor diferencia de electronegatividad, también es más alto el punto de ebullición.
Otro efecto importante es sobre la solubilidad, pues las sustancias que pueden formar puentes de hidrógeno con el agua (alcoholes, por ejemplo) son solubles en ella, siempre y cuando su tamaño no sea muy grande.
Las interacciones dipolo-dipolo se da entre moléculas polares como el HCl o PCl3, por ejemplo. Son similares a los puentes de hidrógeno, pero menos intensas.

Las interacciones dipolo-dipolo inducido se dan en mezclas de sustancias y por ello no las consideraremos aquí.
Finalmente, las interacciones dipolo instantáneo-dipolo instantáneo o de London son universales y se producen como consecuencia del movimiento caótico de los electrones de las moléculas, que, en un instante dado, producen una separación de carga que resulta en un pequeño dipolo que interacciona con los demás que se generan en otras moléculas. A pesar de ser las más débiles de las interacciones, son importantes en bioquímica, debido a que las fuerzas de London aumentan con el tamaño y las biomoléculas son muy grandes. También son responsables del aumento del punto de fusión de sustancias apolares como los hidrocarburos, los halógenos o los gases nobles.



El término cromatografía hace referencia a un conjunto de técnicas empleadas para separar mezclas que incluyen la interacción de los componentes de la mezcla con dos fases: una estacionaria que no se mueve, y otra móvil o eluyente que se traslada a través de la estacionaria. Los componentes de la mezcla se separan en función de su distinta afinidad por las fases estacionaria y móvil. Si el componente interacciona fuertemente con la fase estacionaria, se desplazará poco por ella, y si por el contrario por lo que tiene afinidad es por la móvil, se moverá mucho con ella.
En función de la fase estacionaria y la móvil, resultan las distintas técnicas cromatográficas:
- Cromatografía en papel: la fase estacionaria es papel, en la que se deposita la mezcla, generalmente en forma de una gota de disolución, la fase móvil es un líquido o mezcla de líquidos que se desplaza por el papel por capilaridad

- Cromatografía en capa fina: similar a la anterior, pero la fase estacionaria es una fina capa de sílice depositada sobre un soporte, generalmente de aluminio

- Cromatografía en columna: la fase estacionaria es un sólido que se deposita en un depósito tubular (puede usarse una bureta), la mezcla se deposita en la parte superior de la columna y el eluyente se va dosificando poco a poco desde el mismo punto, desplazándose por la columna por gravedad

- Cromatografía de gases: se parece a la de columna, pero en este caso, la fase móvil es un gas que se introduce a presión por la columna junto a la mezcla, que puede ser gaseosa o líquida, aunque en este caso, debe vaporizarse
- Cromatografía líquida de alta presión: es una mezcla de las dos anteriores, usando como fase móvil un líquido que se introduce en la columna a presión, haciendo el proceso más rápido que en la cromatografía en columna






En las primeras técnicas, la identificación de las sustancias se suele hacer mediante la determinación del factor de retención Rf, que es el cociente entre la distancia viajada por el componente de la mezcla y la distancia viajada por el eluyente. Existen tablas de estos valores para sustancias, eluyentes y fases estacionarias típicas. Para hacer las mediciones es preciso "ver" las marcas de los componentes, para lo que hay varias alternativas: hacer reaccionar las sustancias con un agente revelador (ácido sulfúrico, ninhidrina) o usar soportes sensibles al ultravioleta.



Cuando tratamos con especies con enlaces múltiples, es posible que existan varias opciones respecto a dónde colocar tales enlaces. Ese fenómeno de deslocalización de los electrones se llama resonancia y cada una de las representaciones que tratan de describir a la especie química, forma de resonancia. No debe interpretarse que la molécula es a veces una cosa y a veces otra, o que algunas moléculas son de un tipo y otras de otra, la resonancia trata de representar que los enlaces no son ni simples ni múltiples sino intermedios, pero siendo todos los implicados idénticos entre sí e indistinguibles (ver otras explicaciones en esta WEB).

En esta WEB podéis ver más ejemplos y ejercicios al respecto de las estructuras Lewis y la resonancia.
El ejemplo más conocido de resonancia es el del benceno, C6H6, que en química orgánica se denomina aromaticidad:


Hay pruebas físicas y químicas que apoyan este hecho:
- Pruebas físicas:
- Las distancias C-C son todas idénticas e intermedias entre el enlace simple y el doble

- Los seis átomos de H dan una única señal el 1H-RMN a un desplazamiento químico muy diferente del de los H de alquenos
- No se observa la banda de absorción en IR a 1650 cm-1 típica de los alquenos
- Pruebas químicas:
- No decolora el agua de bromo, cosa que sí hacen los alquenos, indicando que no hay dobles enlaces
- No da reacciones de adición (típicas de alquenos), sino de sustitución en los H
- Los derivados 1,2 sustituidos son un único compuesto, no presentan isomería como cabría esperar si tuviera enlaces dobles alternados
- La entalpía de hidrogenación del benceno es menos negativa que la que correspondería a tres enlaces dobles, dado que la deslocalización de los electrones aumenta su estabilidad


En la práctica, el octete electrónico sólo se cumple estrictamente en elementos del primer periodo, concretamente C, N, O y F; en el caso del B o el H, el octete electrónico no puede alcanzarse por déficit de electrones; y en el resto de los elementos, es posible superarlo ampliamente (esto se llamó expansión del octete, pero esa es una idea obsoleta hoy en día). La razón para ello radica en que a partir del tercer periodo, los elementos poseen orbitales d disponibles para participar en el enlace, lo que permite la formación de más de 4 enlaces:


En estas especies electrónicas, la TRPEV se aplica del mismo modo que en las anteriormente vistas.

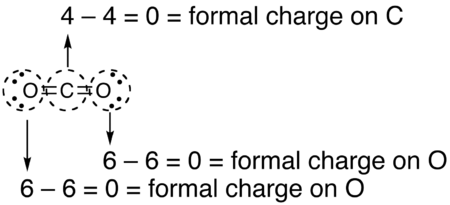
La carga formal se define como la carga eléctrica que tendría el átomo si los electrones compartidos se repartieran a partes iguales entre los átomos que los comparten. En términos de estructuras de Lewis, las estructuras que menos carga formal tengan son una mejor representación de la especie electrónica, aunque ello implique incumplir la regla del octete.


En términos modernos, y en el marco de la mecánica cuántica, en la que los electrones vienen descritos por las funciones de onda, el enlace entre dos elementos se da cuando los orbitales atómicos solapan en el espacio, lo que, a efectos matemáticos, significa que en lugar de estar descritos por la función de onda de un átomo, pasa a estarlo por una combinación lineal de las funciones de onda de los orbitales que solapan de cada átomo:

Cuando el enlace se produce de modo frontal entre orbitales s o p, el enlace tiene simetría axial y se denomina enlace s (sigma). si por el contrario, el enlace se produce lateralmente entre orbitales p, el enlace no tiene simetría axial y en lugar de eso posee un plano de simetría y se denomina enlace p (pi).
Así, el hidrógeno posee un enlace s por solapamiento de orbitales s:

Los enlaces s son el "esqueleto" de las moléculas, los enlaces p se superponen a esta estructura, no pueden existir sin la preexistencia de un enlace s. El benceno al que hemos aludido arriba es un buen ejemplo para visualizar estos enlaces:


El uso de los orbitales atómicos no es suficiente para explicar el enlace en la mayoría de las moléculas porque los ángulos de enlace se restingirían en su mayoría 90º y 180º y la realidad es mucho más diversa. Para solventar este problema se diseñó una estrategia que consiste en sustituir los orbitales atómicos puros por combinaciones lineales de orbitales atómicos del mismo átomo, lo que produce nuevos orbitales atómicos que denominamos orbitales híbridos, que se diferencian de los atómicos en que son direccionales, apuntando en direcciones concretas del espacio. Pero hay que dejar claro que la hibridación del átomo se decide a posteriori: en primer lugar se escribe una estructrua de Lewis para la especie; luego se le asocia una estructura VSEPR, que determina la forma, y para esa forma, hay una hibridación asociada que produce orbitales justo en las direcciones de dicha forma. En las tablas de VSEPR de arriba aparecen también las hibridaciones asociadas.


.png)
No hay comentarios:
Publicar un comentario